Gene Activity Score Prediction Tutorial (Hippocampus)
This tutorial evaluates ATAC accessibility imputation on the Hippocampus dataset by comparing the ground truth matrix to CoxFormer imputed results.
The notebook includes:
Metric computation and a radar summary across methods (e.g., PCC, SSIM, RMSE).
Spatial visualization of selected genes to compare spatial patterns between ground truth and imputations.
Spatial annotation of ATAC clusters (e.g., CA3 and GCL) for regional context.
DEG heatmap analysis for predefined regions (CA3 vs GCL) with shared color scaling across methods.
All functions are available in utils/.
0. Configuration
Define the dataset module (e.g., Hippocampus) and set the paths:
DATA_PATH: input data directory (e.g.,meta.tsv,cnts.tsv,locs.tsv)RES_PATH: imputation results and evaluation outputs (e.g.,{tool}_impute.csv,{tool}_Metrics.txt)
[1]:
## import packages
import os
import pandas as pd
import matplotlib.pyplot as plt
os.chdir(os.path.abspath(".."))
from utils.Gene_expression_prediction_utils import CalDataMetric
from utils.Gene_activity_score_prediction_utils import plot_radar, plot_atac_spatial, plot_spatial_scatter, plot_cluster_heatmaps
[2]:
dataset = "Hippocampus"
RES_PATH = f"Result/Gene_activity_score_prediction/{dataset}/"
DATA_PATH = f"Dataset/Gene_activity_score_prediction/{dataset}/"
1. Load ground truth and imputed matrices
This section loads:
meta.tsv: spot-level metadata (e.g., cluster labels and optional pseudotime)cnts.tsv: ground truth gene activity scores matrix (genes × score){tool}_impute.csv: imputed ATAC matrices from different methods
[3]:
meta = pd.read_csv(os.path.join(DATA_PATH, "meta.tsv"), sep="\t", header=0, index_col=0)
gt = pd.read_csv(os.path.join(DATA_PATH, "cnts.tsv"), sep="\t", header=0, index_col=0)
location = pd.read_csv(os.path.join(DATA_PATH, "locs.tsv"), header=0, index_col=0, sep="\t")
impute_our = pd.read_csv(os.path.join(RES_PATH, "CoxFormer-Loc_impute.csv"), header=0).set_axis(gt.index, axis=0)
impute_cor = pd.read_csv(os.path.join(RES_PATH, "correlation_pca-Loc_impute.csv"), header=0).set_axis(gt.index, axis=0)
impute_cox = pd.read_csv(os.path.join(RES_PATH, "coexpression_pca-Loc_impute.csv"), header=0).set_axis(gt.index, axis=0)
impute_txt = pd.read_csv(os.path.join(RES_PATH, "text-Loc_impute.csv"), header=0).set_axis(gt.index, axis=0)
1.1 Metric computation
This section calls CalDataMetric(...) to compute evaluation metrics for imputation quality and writes results to RES_PATH (e.g., {tool}_Metrics.txt).
Example metrics:
PCC and SSIM: higher values indicate better agreement with ground truth.
RMSE: lower values indicate smaller reconstruction error.
[4]:
CalDataMetric(RES_PATH, os.path.join(DATA_PATH, "cnts.tsv"))
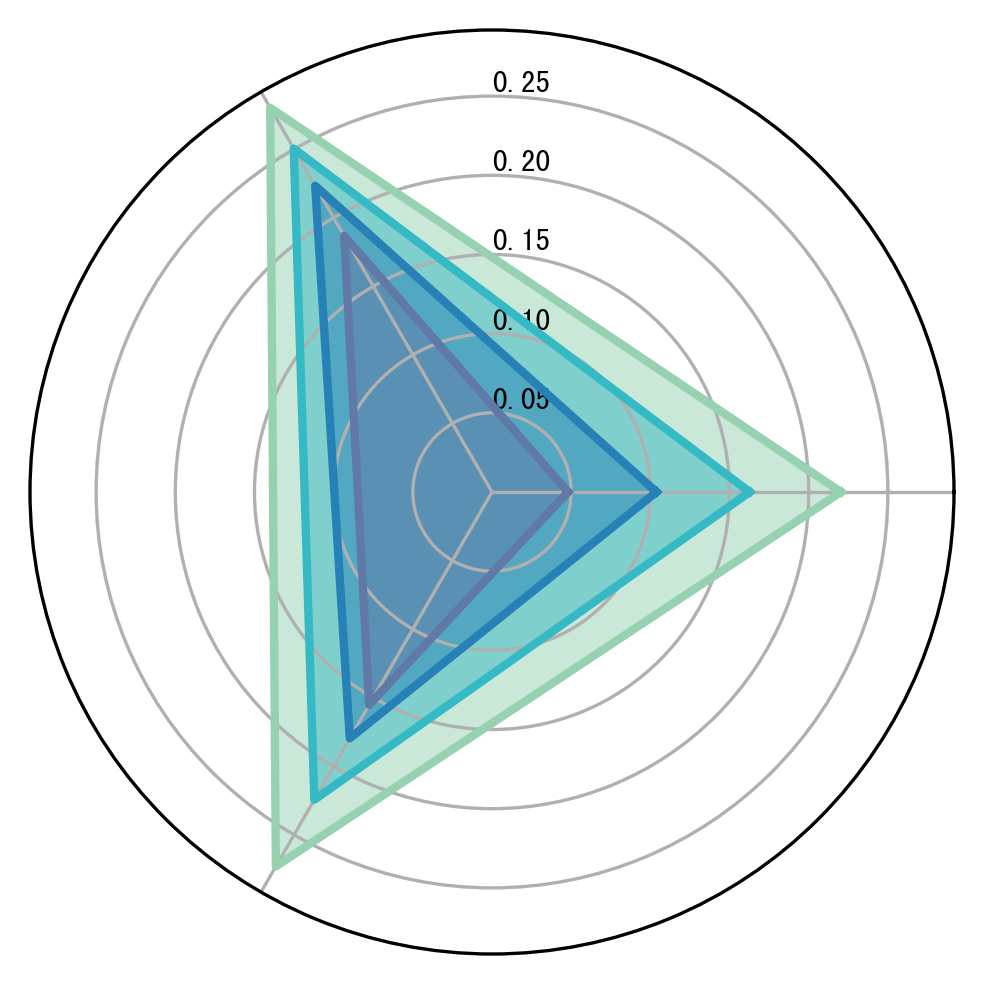
1.2 Radar summary of metrics
This section reads {tool}_Metrics.txt for each method and summarizes the mean metric values across features. A radar plot is used to provide a compact comparison across methods.
Note: RMSE apply a consistent transformation before plotting to avoid misinterpretation.
[5]:
metric = ['PCC', 'SSIM', 'RMSE']
colors = ["#96D2B0","#35B9C5","#2681B6","#6179A7"]
Tools = ['CoxFormer-Loc',"coexpression_pca-Loc","correlation_pca-Loc","text-Loc"]
plot_radar(RES_PATH, metric, Tools, colors, save=False)
2 Selected genes spatial heatmap plot
This section visualizes the spatial patterns of a small set of representative genes. For each selected gene, spatial scatter plots are generated for:
Ground truth
CoxFormer imputation
Co-expression imputation
Correlation imputation
Description imputation
The goal is to compare spatial structure and regional variation across methods under the same visualization settings.
[5]:
Selected_list = ['SMIM27','SMDT1','BIN2','ASAH1','NPAS4']
data = {
"GroundTruth": gt,
"CoxFormer": impute_our,
"Coexpression": impute_cox,
"Correlation": impute_cor,
"Description":impute_txt,
}
plot_atac_spatial(
gene_list=Selected_list,
location=location,
data=data,
save_dir=RES_PATH,
add_colorbar=True,
s=10,
methods=("GroundTruth", "CoxFormer", "Coexpression","Correlation","Description"),
)
Gene: SMIM27
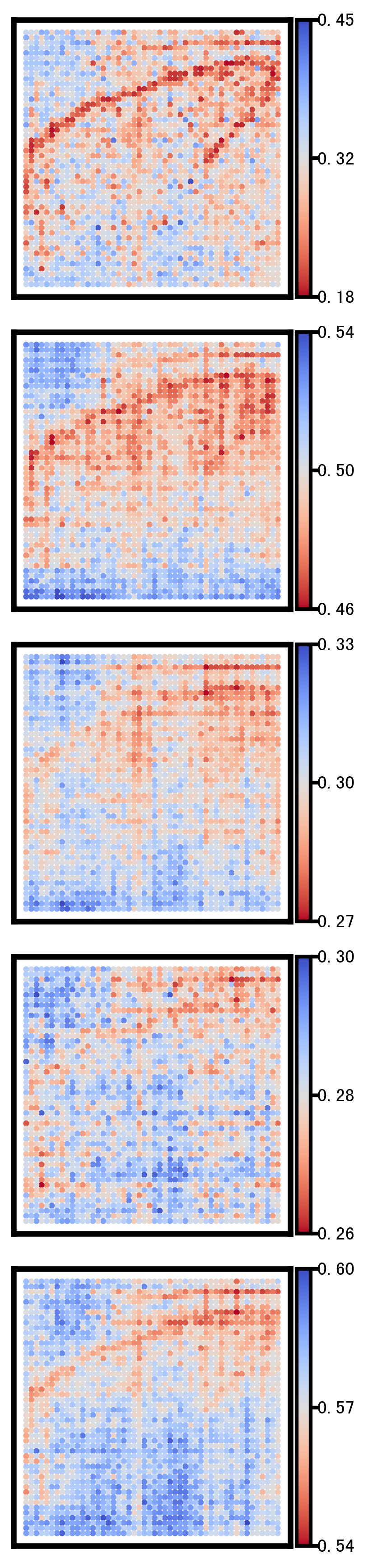
Gene: SMDT1
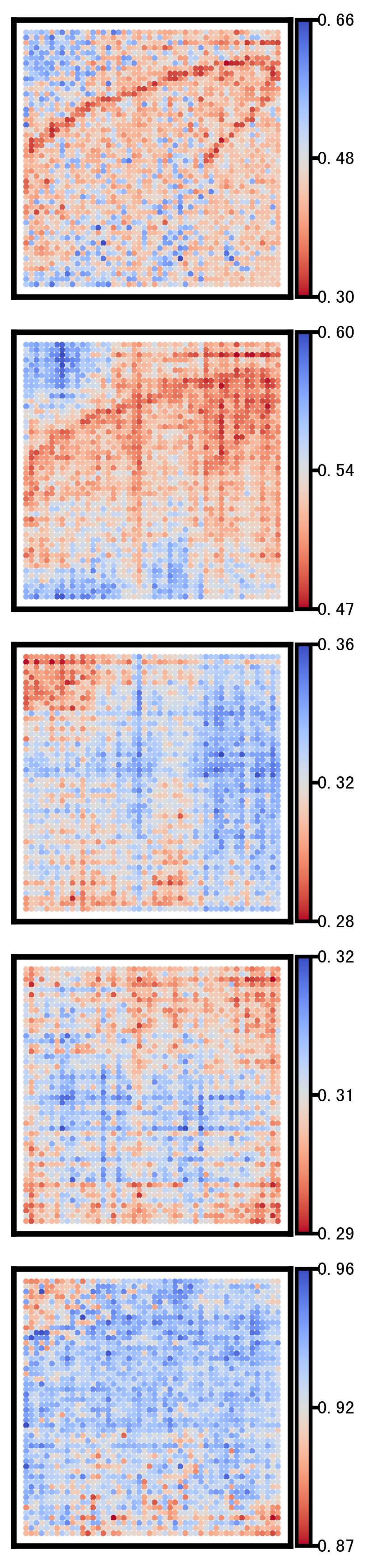
Gene: BIN2
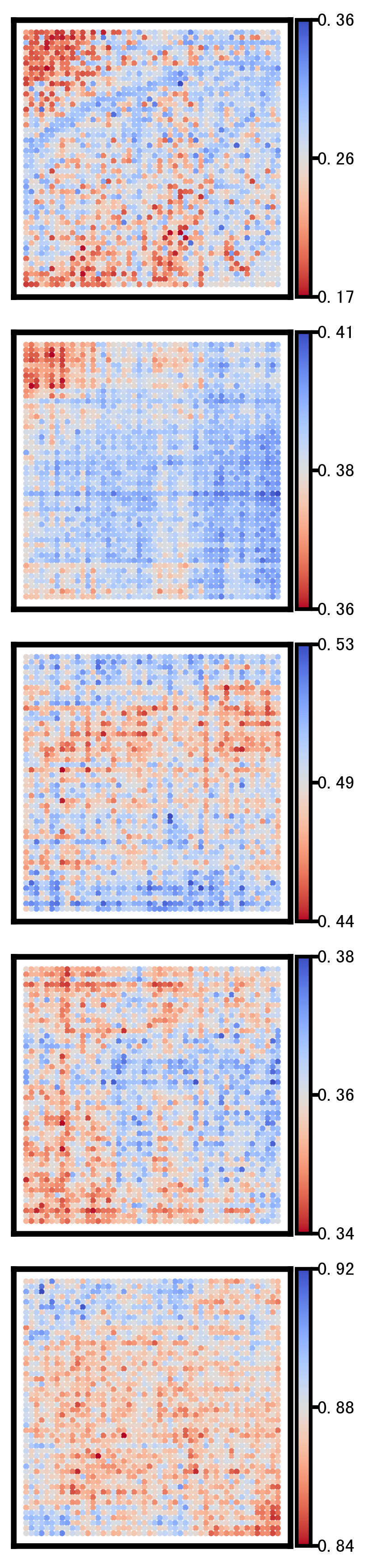
Gene: ASAH1
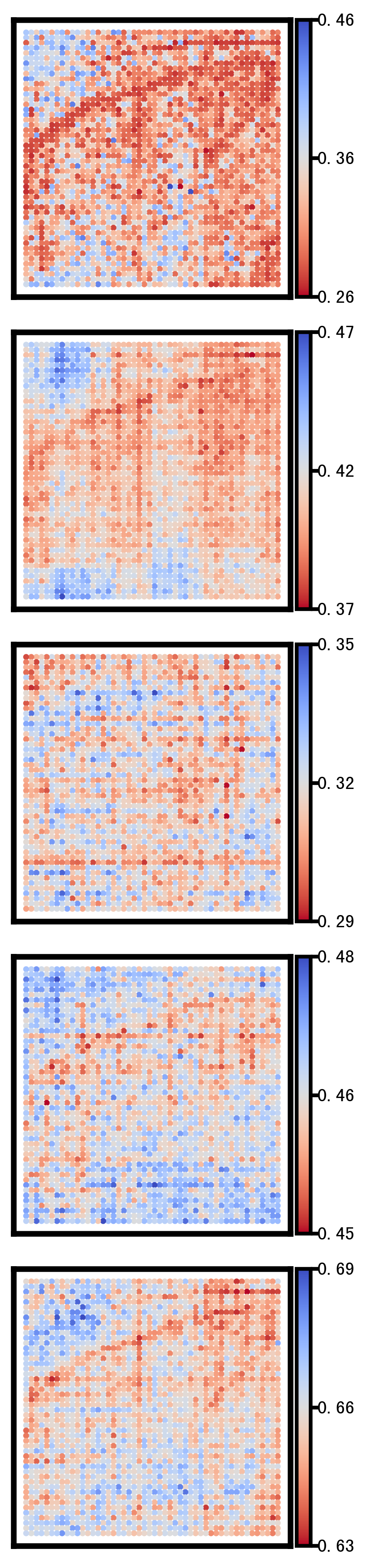
Gene: NPAS4
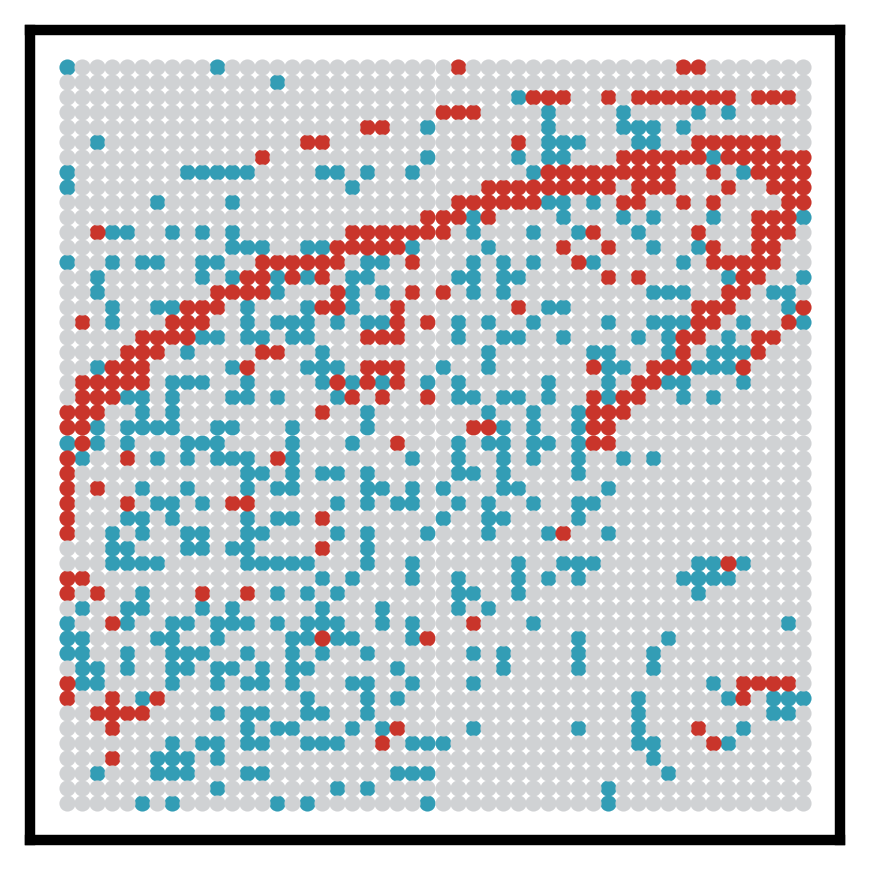
3. Visualize spatial ATAC clusters
This section visualizes cluster annotations from meta["ATAC_clusters"] in spatial coordinates. Cluster labels are optionally mapped to more interpretable region names (e.g., CA3 and GCL), with all remaining labels grouped as “Other”.
This plot provides spatial context for region-specific analyses performed in later sections.
[6]:
name_map = {"C1": "CA3 pyramidal layer (CA3 Pyr)", "C4": "Granule cell layer (DG GCL)"}
color_map = {"CA3 pyramidal layer (CA3 Pyr)": "#339DB5",
"Granule cell layer (DG GCL)" : "#C9352B",
"Other" : "#D0D2D4"}
cluster_series = meta["ATAC_clusters"].reindex(location.index).map(name_map).fillna("Other")
fig, ax = plt.subplots(1, 1, figsize=(3, 3), dpi=300)
plot_spatial_scatter(ax, location, cluster_series, mode="categorical",
color_map=color_map,add_legend=False, s=15)
plt.tight_layout()
plt.show()
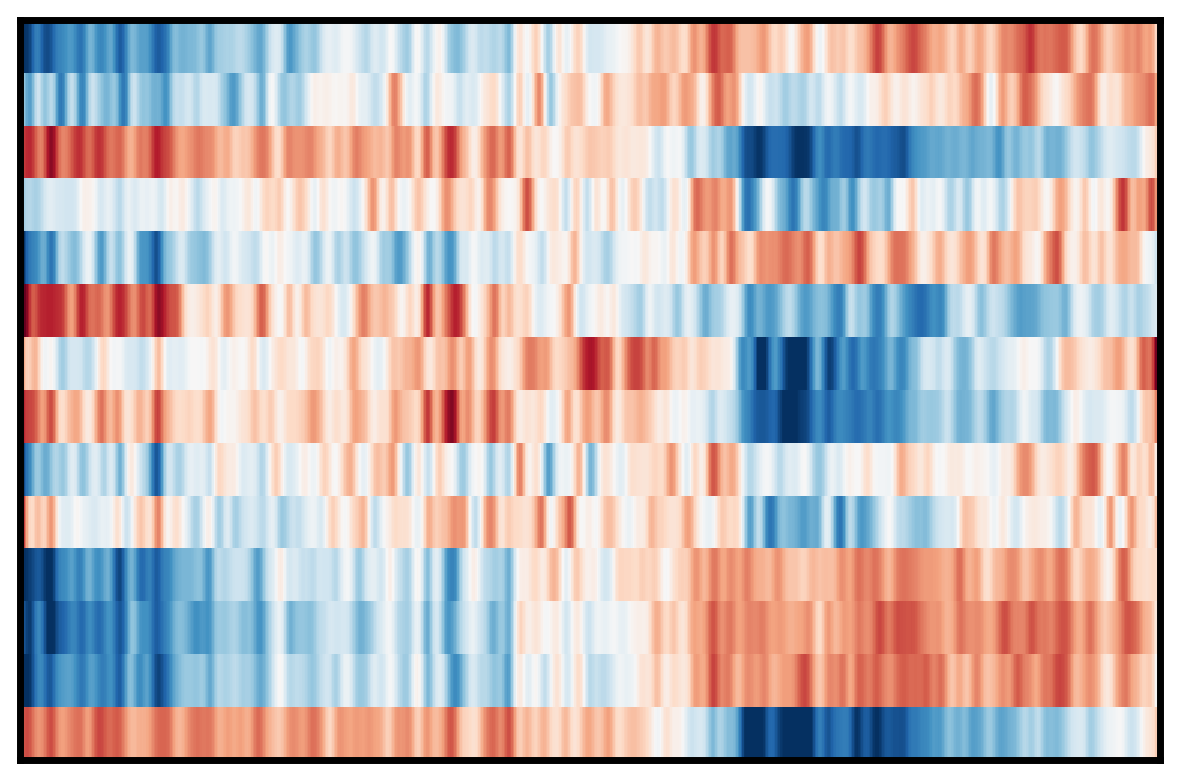
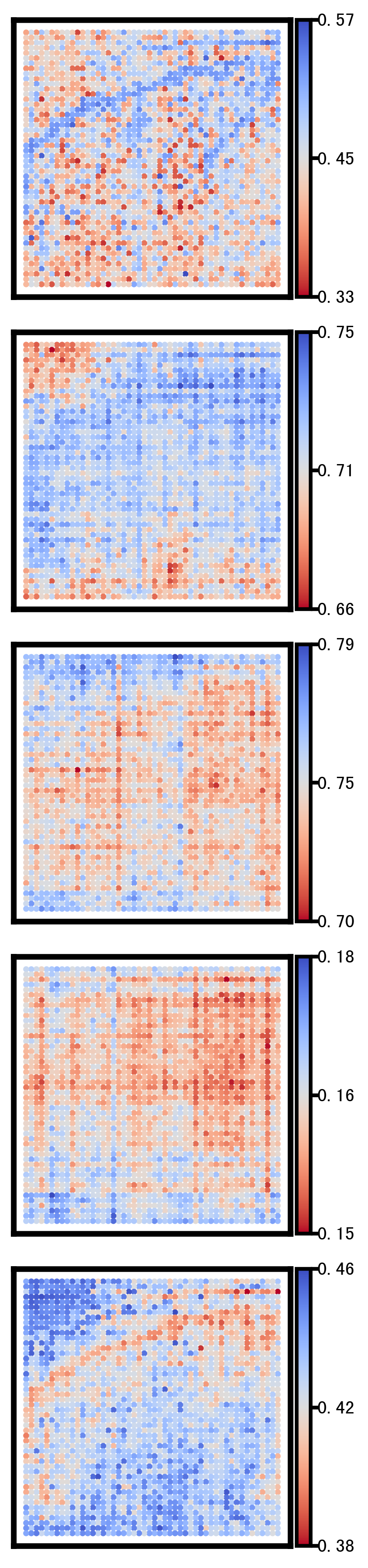
4. Plot DEG of defined cluster
This section compares region-specific accessibility patterns between CA3 and GCL:
Align shared features across ground truth and imputed matrices.
Subset spots to CA3 and GCL based on metadata labels.
For each region, perform a Wilcoxon-based differential analysis to identify top marker features (topK).
Order spots within each region using the mean accessibility of region-specific markers.
Plot heatmaps for ground truth, CoxFormer, Co-expression, Correlation and Description using a shared color scale (derived from ground truth).
This provides a direct visual comparison of region-specific signal patterns across methods.
[ ]:
data = {
"GroundTruth": gt,
"CoxFormer": impute_our,
"Coexpression": impute_cox,
"Correlation": impute_cor,
"Description":impute_txt,
}
plot_cluster_heatmaps(data=data,meta=meta, clusters=["CA3", "GCL"],RES_PATH=RES_PATH)
Ground Truth:
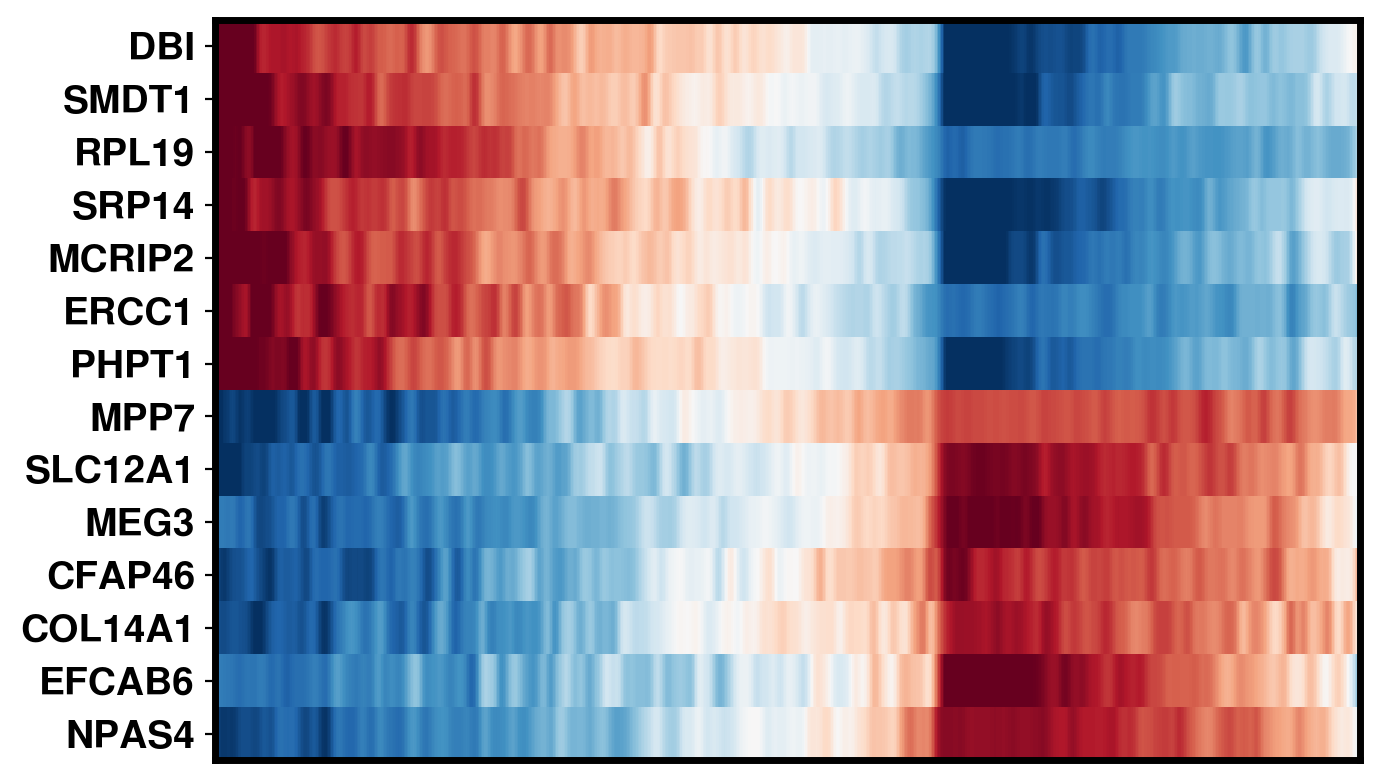
CoxFormer:
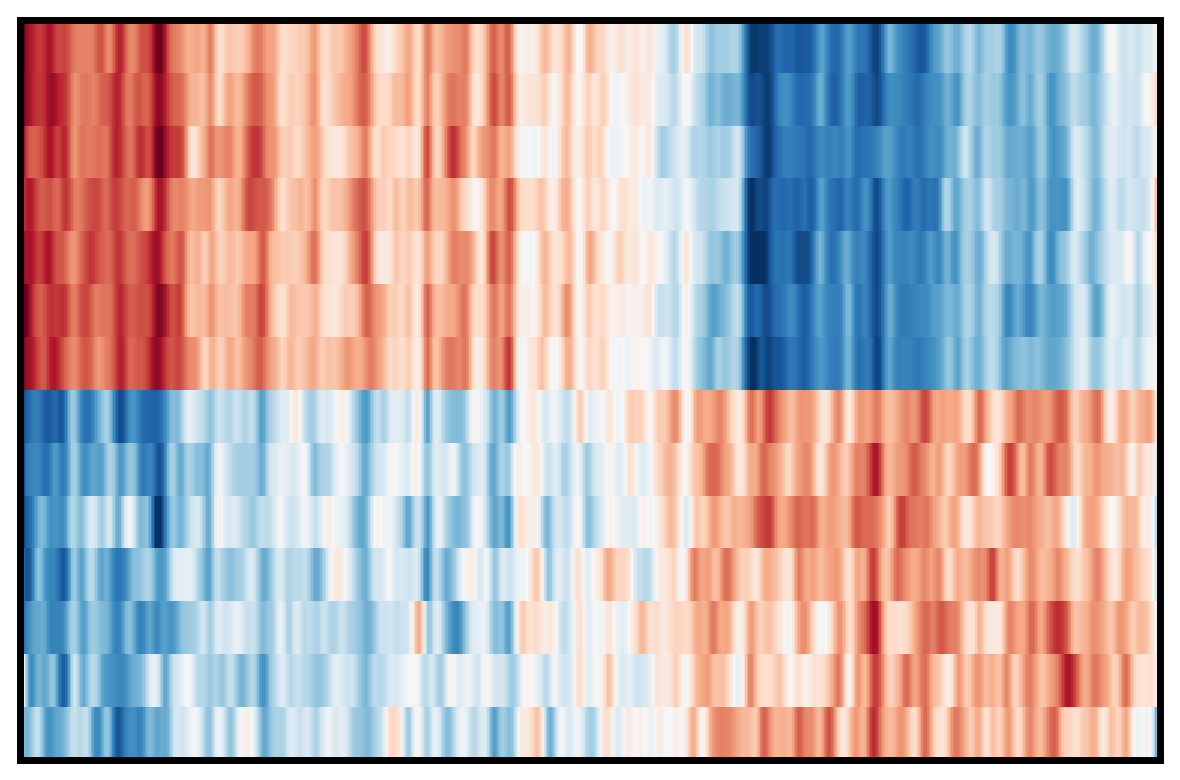
Coexpression:
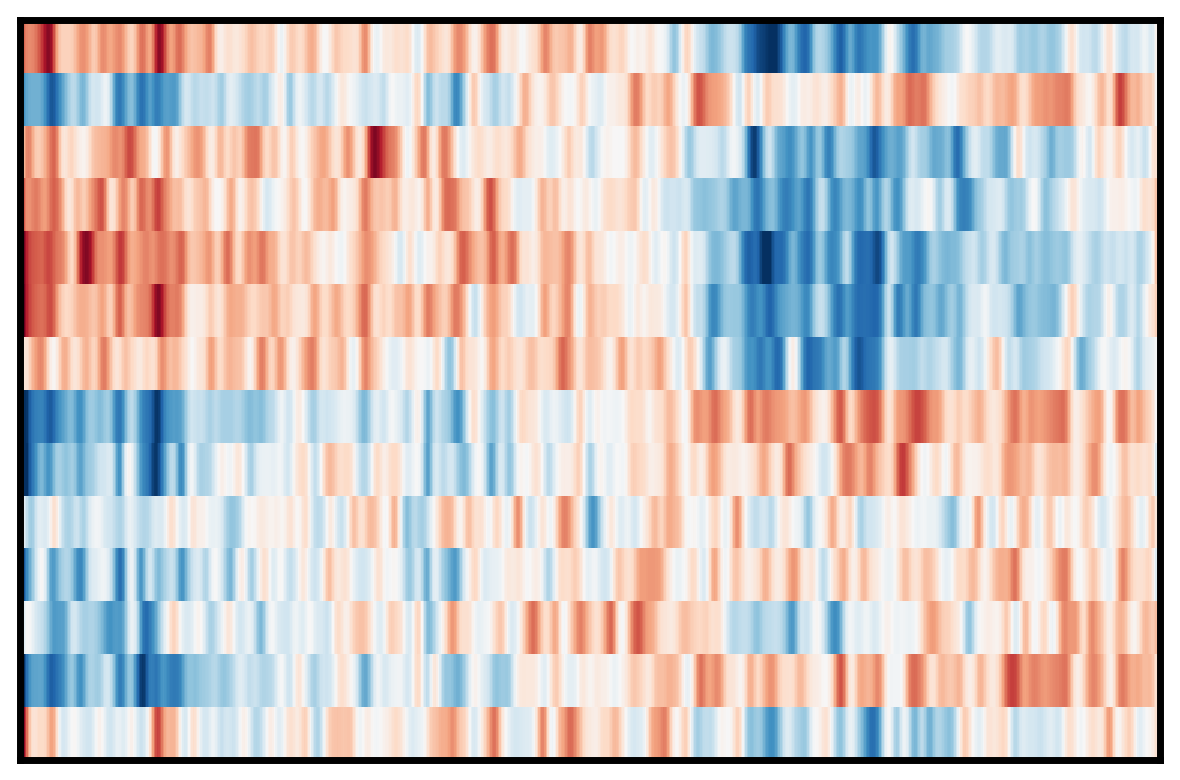
Correlation:
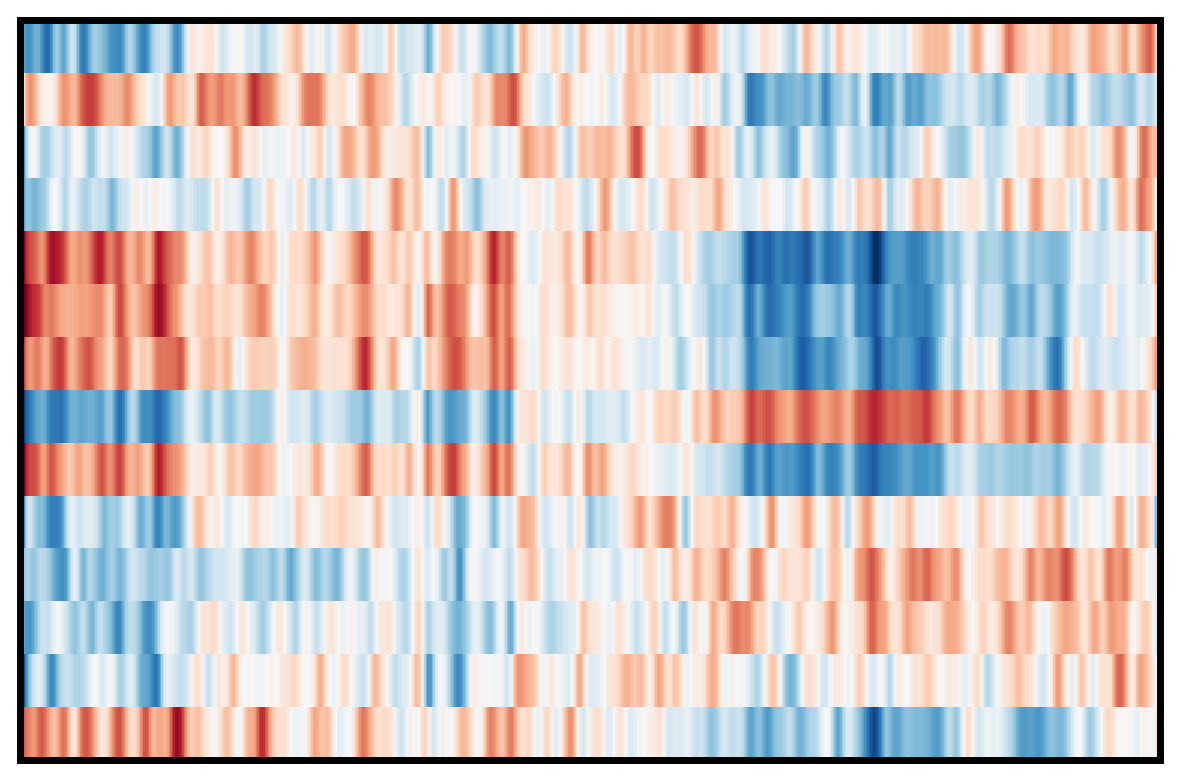
Description: